How Skin Barrier Function Works
What the Skin Barrier Actually Is and What It Does Most people, including a lot of people working in skincare professionally, carry around a mental model of the skin barrier as…
What the Skin Barrier Actually Is and What It Does
Most people, including a lot of people working in skincare professionally, carry around a mental model of the skin barrier as something passive: a film sitting on the surface, something to moisturize, occasionally strip and rebuild. I held some version of that model early on. You work with the actual biology long enough and the wrongness of it becomes impossible to ignore, and costly in practice.
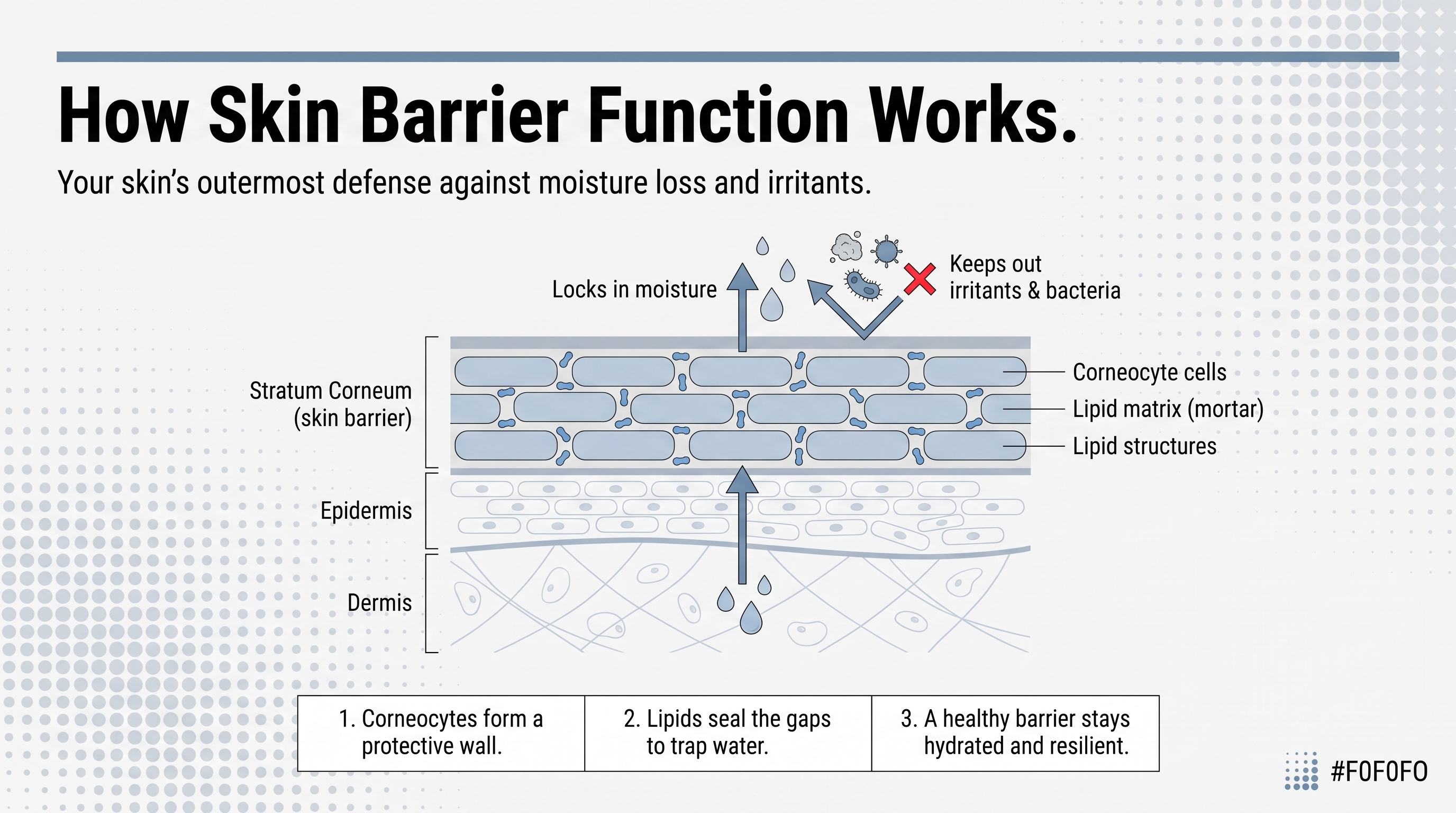
The skin barrier is a chemically active, self-regulating biological system. The outermost layer doing the heavy lifting is the stratum corneum, and its two primary jobs sound deceptively simple: keep water in, keep damaging things out. UV radiation, microbes, physical trauma, chemical insult, temperature extremes. A continuous sheet of tissue sitting between your internal biology and everything the world throws at it.
But what if the barrier is doing considerably more than that? Here is the piece that actually reorients how you think about all of it: the stratum corneum is not static. It is continuously produced through terminal keratinocyte differentiation, where living cells in the lower epidermis mature, flatten, and reorganize into the SC's architecture. The barrier is being built and dismantled simultaneously, which is precisely what makes it repairable, and also what makes it so vulnerable when the conditions supporting that process break down. It also participates in biological signaling, initiating inflammatory responses when it detects breach. Passive wrapper is the last thing it is.
The Brick-and-Mortar Architecture: Corneocytes and the Lipid Matrix
The metaphor the field settled on decades ago still holds: bricks and mortar. I have not found a better one, and I have looked.
Corneocytes are the bricks. Flattened, protein-dense, mechanically tough. They absorb UV, resist physical stress, and regulate hydration within the cell interior. But the bricks alone do not constitute the permeability barrier. The mortar does.
Between every corneocyte sits an extracellular lipid matrix organized into layered lamellar structures, alternating hydrophilic and hydrophobic regions that physically resist water diffusion. The composition runs roughly half ceramides, about a quarter cholesterol, and a smaller fraction of free fatty acids. The ratio matters as much as the presence. Disrupt the proportion between those components and you impair the barrier even if total lipid concentration is unchanged. That distinction is one the product side of this industry has been genuinely slow to internalize.
One more structural detail worth sitting with: the outer lipid envelope of each corneocyte is covalently bonded to the protein scaffold of the cell itself. The mortar is literally attached to the bricks. That raises an important question: if topical water cannot dissolve a covalent bond, what exactly is it doing? It addresses neither the lipid matrix controlling movement across the SC nor the covalently bound envelope connecting that matrix to the cellular architecture beneath. You are simply not reaching what needs reaching.
How Ceramides Are Made and Why Their Variety Matters
Ceramides dominate the lipid matrix, and how they actually get there explains why they are so difficult to replace from the outside.
Ceramide biosynthesis does not happen in the SC itself. Living keratinocytes in the stratum granulosum, the layer just below, package precursor lipids into structures called lamellar bodies, which are secreted into the extracellular space at the granulosum-SC junction. Enzymes there, particularly beta-glucocerebrosidase, convert those precursors into active ceramides. A precision operation running continuously beneath skin you cannot see or feel doing anything. It is remarkable when you stop to consider it.
What comes out is not a single ceramide. There are at least sixteen isoforms in the stratum corneum, each varying by fatty acid chain length and sphingoid base structure, each contributing differently to the density and arrangement of the lamellar bilayers.
This is where the clinical picture gets complicated in ways the skincare industry reliably flattens. In psoriasis, the problem is not simply low total ceramide content; it is a disrupted ceramide subclass composition. The isoform profile is altered. Adding back generic ceramide does not restore that distribution. One might argue that "add ceramides" sounds precise enough — but does it hold up? Not without asking which ceramides, in what proportion, and whether they can organize correctly into lamellar structures once applied. Those are harder questions. The answers are still being worked out, and I think some brands would rather not ask them.
Filaggrin's Role in Hydrating the Corneocyte From Within
The mortar controls water movement between cells. Inside the cells themselves, the story belongs to a protein called filaggrin, and it does two sequential jobs that are easy to conflate.
First, during terminal differentiation, filaggrin aggregates keratin filaments inside the keratinocyte, collapsing and flattening the cell into the compact form the corneocyte needs to be. Second, and this is the part that gets glossed over consistently, filaggrin is then broken down into a collection of small molecules called natural moisturizing factor, NMF. Free amino acids, pyrrolidone carboxylic acid, urocanic acid, urea. NMF is hygroscopic; it pulls water in and holds it. When NMF is sufficient, the corneocyte stays plump and structurally cohesive. When it is not, cells shrink and intercellular gaps form, creating physical pathways for water loss.
It is also worth considering one detail here that connects directly forward: filaggrin catabolism, that breakdown into NMF, requires an acidic environment. The enzymatic process operates optimally around pH 5.5. If pH rises, filaggrin is not broken down efficiently, NMF production falls, and hydration suffers. The chemistry of the corneocyte interior is pH-dependent, which means the acid mantle is not just a surface phenomenon, something I will come back to.
Loss-of-function mutations in the filaggrin gene are among the most well-established genetic risk factors for atopic dermatitis. The cascade they set off is one of the clearest demonstrations in all of dermatology of how a single structural deficit propagates system-wide failure: reduced NMF, impaired hydration, altered pH, disrupted ceramide production, compromised barrier, sensitization to environmental allergens. One protein, and everything downstream shifts.
The Acid Mantle: How Surface pH Orchestrates Barrier Chemistry
The term "acid mantle" was coined in 1928, which makes it one of the older concepts in skin science still in active clinical use. Adult skin surface pH sits around 5.0, within a healthy range of roughly 4.0 to 5.8. Slightly acidic, and notably so compared to body fluids. That acidity is not incidental.
To understand why this works, we must first look at the pH gradient across the SC's depth, and how that gradient is regulating multiple biological processes at once. Deeper in the SC, where pH is lower, a serine protease inhibitor called LEKTI stays bound to its target enzymes, holding them inactive. These enzymes, KLK5, KLK7, and KLK14, when active, break down the protein structures holding corneocytes together, enabling controlled cell shedding. As corneocytes migrate outward and pH rises slightly toward the surface, LEKTI releases, the enzymes activate, and orderly turnover occurs. Disrupt the gradient, and you disrupt the timing of shedding in ways that are not always visible until the problem is well established.
Antimicrobial peptides are also pH-dependent. Dermcidin shows bactericidal activity against Staphylococcus aureus that drops from greater than 90% efficacy at pH 5.5 to around 60% at pH 6.5. One pH point, nearly a third of your antimicrobial defense.
And then there is beta-glucocerebrosidase, the enzyme responsible for converting precursor lipids into active ceramides. It is acid-preferring. Alkaline shifts impair its function, meaning an elevated skin pH simultaneously undermines lipid production and antimicrobial defense. The acid mantle orchestrates the barrier's entire chemistry. When I see a product formulated at pH 8 marketed as "gentle," that is what I keep thinking about.
Tight Junctions: The Living Epidermis Adds a Second Line of Restriction
Below the stratum corneum, in the viable epidermis, there is a second structural barrier that does not receive nearly the attention it deserves. Tight junctions.
These are protein complexes, claudins, occludin, zona occludens among the key players, that form a belt around epithelial cells and seal the paracellular space. They restrict the outward movement of water and water-soluble substances through the living epidermis, acting as a backup to the lipid matrix above. They also support keratinocyte behavior during wound healing, including proliferation and differentiation, so they are not simply passive seals.
There is an immune dimension here too. Langerhans cells, the skin's resident antigen-presenting sentinels, are embedded in this same epidermal layer. The tight junction zone is an active sensing interface, where the physical state of the barrier connects directly to immune surveillance.
Microbial lysates from Lactobacillus and Bifidobacterium species have been shown to enhance tight junction protein expression. Staphylococcus aureus disrupts it. In atopic dermatitis, where the stratum corneum is compromised, allergens reach Langerhans cells through weakened tight junctions and drive sensitization. The structural failure and the immune dysregulation that follow are not sequential events. They are concurrent, each accelerating the other.
The Skin Microbiome as a Functional Part of Barrier Maintenance
Staphylococcus epidermidis, the commensal workhorse of healthy skin, produces an enzyme called sphingomyelinase that converts host sphingomyelin into ceramides. A bacterium living on your skin surface is actively contributing to the lipid mortar of the barrier. I sat with that for a while the first time I encountered it. It reframes what "skin health" even means. S. epidermidis also secretes molecules that dampen pro-inflammatory cytokine production and support wound healing, meaning it contributes to both the structural and immunological dimensions of barrier maintenance, simultaneously.
Staphylococcus aureus does essentially the opposite. It disrupts tight junction integrity, colonizes a substantial majority of atopic dermatitis patients, and drives inflammatory flares through toxin production and immune activation. The shift in Staphylococcus balance from epidermidis to aureus is both a symptom of barrier compromise and an accelerant of further damage, at the same time.
But how does this affect our original premise that the barrier is something you apply products to? The field has moved well past treating the skin microbiome as cosmetically interesting. It feeds directly into ceramide production, immune tone, and physical coherence. Commensal composition is a functional parameter, not a wellness amenity. Treating it as the latter shapes which products you trust and which questions you forget to ask.
What Barrier Failure Looks Like and Why It Compounds Itself
The clinical marker most commonly used to assess barrier function is transepidermal water loss, TEWL: the rate at which water diffuses from deeper skin layers to the surface and evaporates. Elevated TEWL signals that the lipid matrix and tight junction systems are underperforming. In atopic dermatitis, skin pH rises to 6.0 or higher even in non-lesional areas. That alkaline shift impairs ceramide biosynthesis, increases serine protease activity beyond what the gradient was designed to produce, and reduces antimicrobial peptide efficacy, all at once.
What makes barrier failure clinically difficult is not that it involves multiple systems. It is that those systems loop back on each other. Lipid failure, pH dysregulation, and microbial imbalance are not parallel problems running on separate tracks. They are the same problem cycling through different components, each pass lowering the threshold for the next.
Th2 cytokines, the dominant immune signature in atopic dermatitis, suppress the expression of multiple barrier genes: filaggrin, loricrin, claudin-1, aquaporin-3, involucrin. Structural failure activates an immune response that then suppresses the genes needed to rebuild the structure. The barrier's integration is its greatest strength and precisely what makes disruption self-reinforcing.
Psoriasis presents its own version of this. The current understanding has shifted toward treating barrier impairment not as a downstream consequence of psoriasis but as an early pathogenic driver. The assumed sequence, inflammation first, structural damage second, is considerably more bidirectional than that. The same revision of assumptions is warranted in conditions we have not studied with the same rigor.
How Barrier Integrity Is Measured and What the Numbers Mean
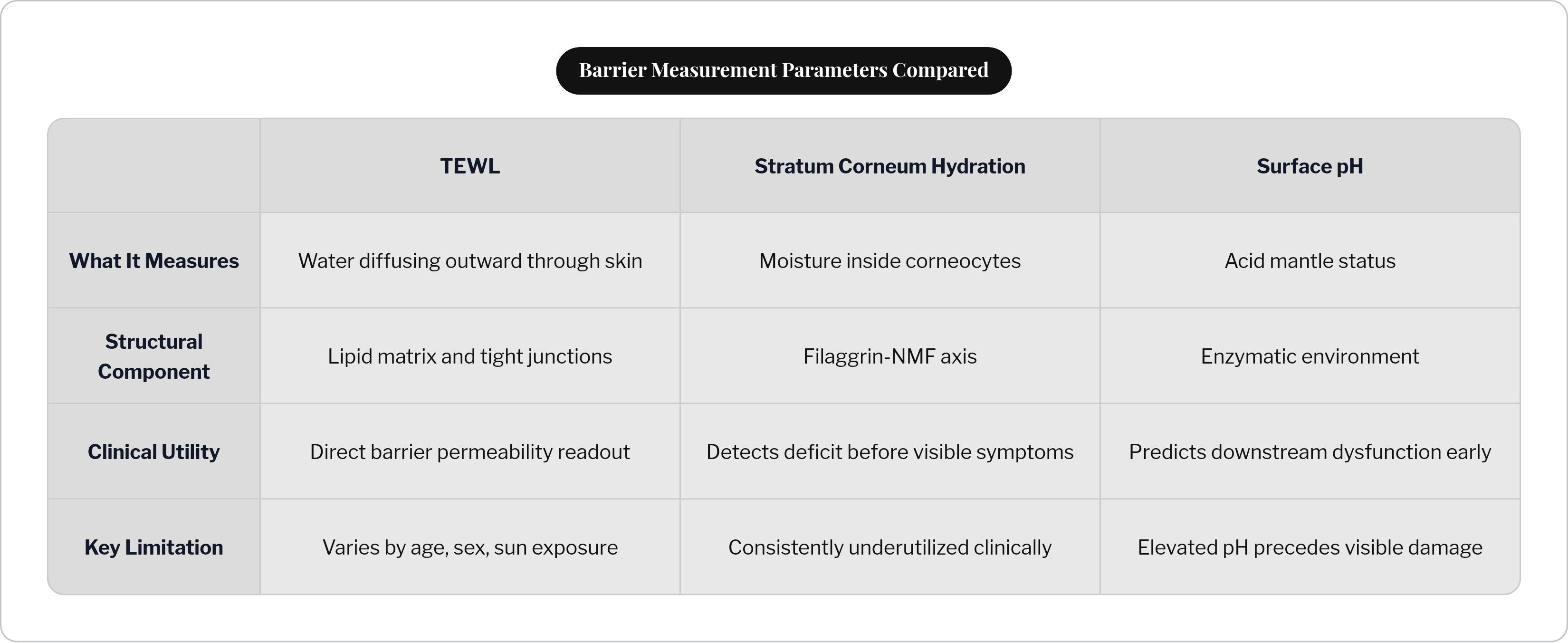
Clinical assessment of barrier function involves several standard parameters, each mapping onto a specific structural component.
TEWL measures the flow density of water diffusing outward from the dermis and lower epidermis to the skin surface, a direct readout of how well the lipid matrix and tight junctions are containing water. Stratum corneum hydration, SCH, reflects what is happening inside the corneocyte, telling you whether the filaggrin-NMF axis is functioning. Low SCH appears before any visible symptoms, which is clinically useful and consistently underutilized. Surface pH measurement assesses acid mantle status directly, and elevated pH predicts downstream enzymatic dysfunction before it manifests as anything visible.
TEWL and SCH together earn the most attention because they map onto distinct structural components: one to the lipid and tight junction systems, one to the filaggrin-NMF axis. Two numbers, two different parts of the architecture. The difference between them shapes what you do next.
Baseline values also differ across individuals by age, sex, and sun exposure history. TEWL is simultaneously a disease marker and a biological variable. The number alone is not the answer; context determines what it means.
Repairing the Barrier by Targeting Its Specific Components
The structural picture built across this piece is not just descriptive. It functions as a diagnostic framework, and that is where it actually earns its keep.
Topical ceramide formulations are proposed to re-establish damaged lamellar architecture, not simply add moisture or occlusivity. Whether applied ceramides are functionally incorporated into the existing matrix depends on their isoform composition and their capacity to self-organize into lamellar structures. Research in cosmetic science has been increasingly focused on exactly this question. The answer so far is that isoform profile and formulation architecture matter considerably. A ceramide cream that deposits lipids on the surface without integrating into the SC matrix is doing something, but not that.
NMF-targeting approaches work through an entirely different mechanism, addressing hydration inside the corneocyte rather than the extracellular space. Humectants like urea, amino acids, and pyrrolidone carboxylic acid are NMF components or functional analogs. They address a different deficit than ceramide deficiency, and confusing the two is more common than it should be.
Slightly acidic cleansers and topicals formulated below pH 5.5 are not a cosmetic preference. Normalizing surface pH restores the enzymatic conditions ceramide biosynthesis requires and antimicrobial peptide activity depends on. A cleanser formulated at pH 8 is not mildly suboptimal. It is actively dismantling the orchestrating condition for multiple barrier processes at once.
Microbiome-aware formulations follow the same logic. Supporting commensal populations, or at minimum not indiscriminately disrupting them, is consistent with how the microbiome actively maintains both the lipid layer and immunological equilibrium.
So the question worth asking of any barrier-focused product is not "does this moisturize." It is: which component is this targeting, at what layer, through what mechanism, and does the formulation have what it needs to actually reach that layer. Those questions do not always yield satisfying answers in the current literature, and some of the most confidently marketed products fall apart quickly under that kind of scrutiny. But the questions themselves are clarifying, and in my experience, asking them is the work.